Creating a new plugin: 1
For this article, I'm going to do something a little different and work through the development of a new plugin as I do it. Not once it's all complete and working but the steps I follow as it progresses, complete with mistakes and problems. I'm not going to detail every line of code, of course, but the steps to be taken and how they are made to work, complete with code snippets where appropriate.
In doing this my hope is twofold: that it will be of interest to anyone wanting to see how a plugin is developed and perhaps thinking of giving it a go, and secondly that it will help to organise my thoughts when writing what is likely to be a tricky piece of code.
What is the plugin?
Well, I already have a plugin called 'ChemLoader' which loads a file containing the definition of a chemical compound and creates that compound on screen. This works well, but it only loads files for relatively small molecules; in particular, it does not load protein molecules, which are usually much larger and more complex. So that is the aim: to write a companion plugin to load protein data files and create the protein in a 3D scene.
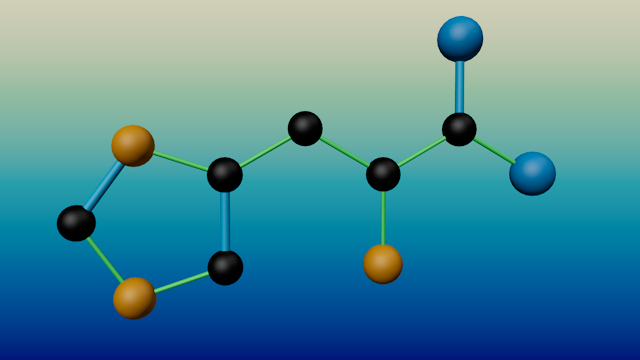
The amino acid histidine (created by ChemLoader)
One thing I do need to point out: I know a little about proteins but not much (and not as much as I thought I did when I started looking into this!). So there may well be errors in my interpretation of protein structure and of PDB files in particular. Hopefully though, it will still make attractive images that some may also find useful. The point is, this is not intended for serious scientific work. It may be useful to some extent but I'm not going to pretend that anyone working with proteins should rely on what it shows.
One plugin or two?
One thing I could do is modify ChemLoader to handle proteins as well - sort of a 'ChemLoader 2.0'. The problem with this is that when testing it I would not only need to test the new code but the existing functions too, because it's all too easy to change something and break functionality that already works. So I decided to create a wholly separate plugin but reuse some of the code from ChemLoader, especially to do with colours and materials, since that already works and it seems pointless to reinvent the wheel. Also, it means that I can reuse some of the interface from ChemLoader, saving time and also making the two plugins look like a complementary pair, rather than completely separate plugins.
Welcome to 'PDBLoader'
That is the name of the new plugin. The first thing was to create the solution file using CMake and use the old source code from ChemLoader. I just needed to get a new plugin ID from Maxon, as otherwise the two plugins would clash and Cinema would object. That done, I now have a copy of ChemLoader with a new name and ID value. It's going to need some interface changes but that can wait until I firm up the additional parameters I will need and remove those that I realise I don't need. The nice thing about this is that I can start inserting some code into the new plugin and build it and test it immediately. Before that though, I need to do some research into the protein data files I'll be reading.
PDB files
The standard format used for describing protein molecules is the Protein Data Bank or PDB file. Actually, there are different versions of this file format but I'm going to use the older or 'legacy' version because there are a lot of these files around and plenty of documentation on the format. You can find lots of these files, and a detailed description of the format, on the PDB website.
PDB files are just text files. They contain a huge amount of information, a lot of which is not relevant for this purpose, but it means loading and parsing a very large text file and abstracting the data needed. That will be the first task: what data do we need, and how to we load it and store it for use?
Protein descriptions
There are problems with proteins that don't exist with smaller molecules. They are made up of chains of amino acids (polypeptide chains) and can be very large. Even a small protein like the hormone glucagon has 247 atoms, not including hydrogen atoms. A protein may be constructed from more than one peptide chain; glucagon has one chain, insulin two, haemoglobin four, and so on. This needs to be taken into account when loading data about individual atoms - you can't simply add them to one long chain. If that wasn't enough, proteins can contain molecules that are not amino acids at all, such as haemoglobin, which contains a molecule of haem as well as four peptide chains. This is a problem when it comes to determining bonds between atoms.
Atoms in a protein molecule
This is the core of any PDB file. Although there is a lot of additional information, the critical data we will need are the data for individual atoms. The file has one line of text in a standardised format per atom. So the very first function we need is one which will load and parse this atom data, because once we have that, we can at the very least display the atoms on screen. Note that most PDB files do not contain details of hydrogen atoms and that is fine - it cuts down the amount of data and simplifies the display. In fact I'm going to take a decision that even if the file does contain hydrogen atom data, I'm going to ignore it. So what does the atom data look like?
Here are the first 10 lines of atom data from a glucagon PDB file (they actually start at line 384 in the file, so there's a lot of mostly extraneous data to read through before we even hit the atom data!):
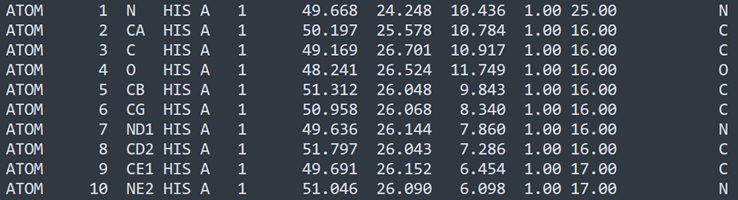
Now what's all this? Taking the first line, we can break it down as shown below. Note that the various fields are located at specific points in the line, so for example, the amino acid identifier ('HIS' in this line) occupies columns 16, 17 and 18. This makes it easy (or should do) to pick out specific data items. Annoyingly, some non-standard usage seems to have crept in, so that the same identifier could be a four-letter code instead of three. Non-standard usage like this will be ignored in this plugin.
1. ATOM: indicates that this line is a line of atom data, so we can ignore (for the moment) all lines in the file beginning with anything else.
2. Numeral 1: this is the number of the atom in the list. You would think it would always start with '1' but it doesn't, for reasons that aren't relevant here. Fortunately we can ignore it as it isn't going to be used by the plugin.
3. Letter N: this is the name of the atom (NOT the atomic symbol). The name is critically important when displaying the bonds between atoms, as we shall see. The first letter of the name is always the atomic symbol, so this is a nitrogen atom (atomic symbol 'N'), but there are two more atoms in this amino acid with the names ND1 and NE2 - but they are still nitrogen atoms.
4. String HIS: the three-letter code for the amino acid this atom belongs to, in this case histidine.
5. Letter A: indicates that this atom is in an amino acid in chain A of the protein, which in this case is the only chain. Other proteins with multiple chains will use different letters to indicate the chain.
6. Numeral 1: indicates that this is the first amino acid in the chain; all the atoms in this molecule of histidine with have this number. All atoms in the next amino acid will have '2' here and so on.
7. Three floating point numbers: the coordinates of the atom in 3D space. We will use these to place a sphere representing the atom into the 3D scene.
8. Numeral 1.00: occupancy (can be ignored by us).
9. Numeral 25.00: temperature factor (can also be ignored by us).
10. Letter N: this is the atom's atomic symbol, so N for nitrogen, C for carbon, O for oxygen and so on. For some reason some PDB files attach a zero to the symbol (e.g. 'C0') which is known to cause problems with protein visualisation software, so we need to account for that when parsing the file.
Not quite that simple...
There are (of course) some caveats to this. First, some ATOM lines contain additional data; for example, immediately after the atom name (item 3 above) there can be a single character to specify alternate locations, and there are other data we can ignore for now.
All we really need are:
- the atom name (important for displaying bonds)
- the amino acid identifier (again, needed for bonds)
- the chain ID
- the coordinates
- the atomic symbol (so we can assign a colour to atoms of the same type)
I think that, for the minute, that is enough. I now need to implement the reading of the PDB file and getting the atom data into a data structure we can use to create the atoms. The next entry in this series will cover that.
Page last updated May 17th 2026