Creating a new plugin: 9
A small (or not so small) problem...
Before looking at disulphide bonds, I found an unexpected problem. On loading a very large file with more than 41,000 atoms the plugin froze, or seemed to do so. I'm not sure it really did, since the molecule was eventually displayed, but some bonds were clearly incorrect, stretching between atoms which were very far apart. What was going on?
It turned out that this molecule contained multiple 'models' - 20 of them to be exact. The reason for this is that with some molecules the structure can have several variants. All have the same chemical structure but the atom locations are different. In such cases, the PDB file will contain a 'NUMMDL' record which simply gives the number of models. The ATOM records are then repeated for each model variant, one after the other - which is why this file was so large. Each model in the file starts with a 'MODEL' record with a number, starting with 1 and incrementing for each model. The ATOM records for each model finishes with an 'ENDMDL' record.
The plugin was behaving correctly and loading all the ATOM records it found, which meant it was loading the same records 20 times and creating the bonds 20 times over, and of course joining all the atoms together in one long chain, since the chain IDs are the same for each model. No wonder it took so long, or that spurious bonds were created. The problem is how to handle this.
Only one model
As the plugin is currently written is is not possible to load all the models but just display one. Therefore, at least for the moment, I've decided to load only the first model in any file containing more than one. This is easy to do: when loading the ATOM records, we just check for the first ENDMDL record and stop loading any more ATOM records at that point. Another way to implement this might be to ask the user which model is to be loaded, and I might do that later on.
Back to disulphide bonds
This turned out to be a little trickier than expected. The SSBOND records (see the previous article) will tell us how many of these bonds exist and I tried several possible methods to find the positions of the bond atoms, but in the end I did it like this.
First, all SSBOND records are stored in a separate array when the file is processed, so in addition to the array of ATOM records we now have a small array of SSBOND records.
Next, we know that these bonds only involve sulphur atoms and occur between cysteine residues. Although most cysteine residues in a chain won't be used to create a disulphide bond, it's easy enough when generating bonds to store all the sulphur atoms in all the cysteine residues in their own array. For this we need a separate struct, like so:
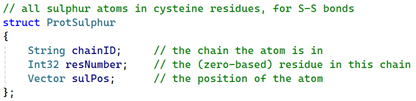
This stores the ID of the chain the residue with a sulphur atom is in, plus the residue sequence number in that chain, and the 3D position of the atom.
Finally, when generating the bonds, in addition to looking for the atoms in peptide links and for the 'OXT' terminating oxygen, we can now look for sulphurs and store them in an array of the above struct. Although this might seem like duplication, it's much easier to search through a small array of sulphur atoms than searching the entire array of ATOM structures for sulphurs. The code is simple:

The only downside is that the array will contain sulphur atoms not used in disulphide bonds, but that doesn't matter since the array will still be a small one.
Creating the spline
In summary we have two arrays: one of the SSBOND records and one for the sulphur atoms in the entire molecule (we can't do this by chain since these bonds are often between different chains). Each SSBOND record will give us, for the two atoms in the bond, the sequence number of the residue containing the sulphur atom, and the chain ID. All we need to do for each SSBOND record is to iterate through the array of sulphur atoms until we find the two which match - that is, which are in the correct residue in the correct chain. So for insulin, for example, the file looks like this:
![]()
So in this case we would parse each of these three records in turn. The first record would tell us that we need residues 6 and 11, both in chain A. Iterating through the array of sulphur atoms would give the 3D position of the two atoms which match those criteria. These are stored in an array of points and once complete this array is processed in exactly the same way as the main set of points to give a spline displaying only the disulphide bonds. This is kept as a separate spline from the main spline so that it can be coloured differerently.
The result
For insulin, the bond display (atoms not shown, for clarity) would look like this:
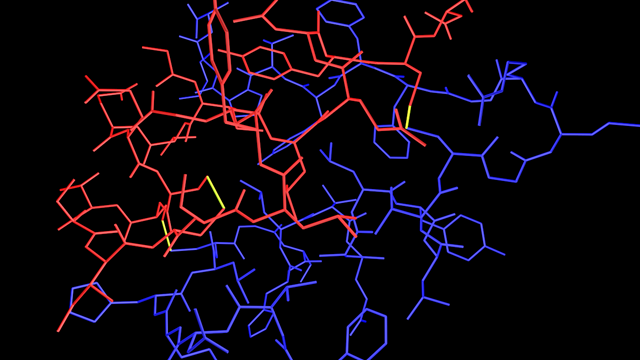
You can see the disulphide bridges clearly shown in yellow. If you look carefully, you can see that one bond is between atoms which are both in the red chain, while the other two are between atoms in the red and blue chains.
For now, that completes the first part of the plugin. The next section will look at handling nucleotide chains instead of polypeptide chains. The principles are the same but there are some subtle differences.
![]() Page last updated June 7th 2026
Page last updated June 7th 2026