Creating a new plugin: 13
Heterogen atoms
So far, the plugin has based creation of the model around the ATOM records. These are used for polymer atoms - that is, those part of a chain of molecules such as amino acids or nucelotides. However, proteins can contain a variety of other atoms or molecules which aren't part of such a chain. These are known as heterogen atoms - 'het atoms' for short - and the PDB file contains these in records similar to ATOM records and known as HETATM records. So, we need to be able to read those records, too.
But that isn't all. There are other record types which we will need in order to create these molecules successfully. These include LINK records (for links between het molecules) and CONECT records (for links within a het molecule, since they don't have standard structures as amino acids or nucleotides do). In addition, there is one HET record for each occurrence of a het molecule and a HETNAM record for each type of molecule! That's a lot of data to keep straight. Fortunately, the structure of a HETATM record is identical to the ATOM record apart from the record identifier so we won't need a different data structure to hold the HETATM records.
We will also have to make a change to the handling of ATOM records. All atoms have a sequence number; this is the number of the amino acid or nucleotide in the chain, and is the same for all atoms in the same amino acid/nucleotide, or indeed in the same het molecule. But in addition each individual atom has its own unique serial number. In glucagon, for example, there are 29 residues so the sequence number ranges from 1 to 29. But there are 246 atoms, so the atom serial number ranges from 1 to 246. Until now we haven't needed that but now we will, because HETATM records also contain atom serial numbers and these are used to link atoms together using a CONECT record. So we will need to read these numbers.
HETATM records
Reading these is easy, because the format is identical to ATOM records, as noted above. One question is whether to create a new array to hold HETATMs or use the existing array we already have for ATOMS. My initial thought was to create a new array, but it soon become clear that this complicated matters and adding them to the existing array was simpler. However, we do need a way to distinguish amino acid/nucleotide atoms from het atoms, so I added a field to the data structure used for each record. This was a simple Bool which was set to true if the atom was a HETATM and otherwise false.
Displaying the atoms is straightforward; the same things regarding 3D position, colours and/or materials apply to them as to the atoms from ATOM records. There is a difference, though. In ATOM records the atoms form part of a molecule - an amino acid or nucleotide - which forms a chain with other such molecules. This is not true, or may not be true, of HETATM records. The atoms specified in these records can be:
- isolated atoms which do not have a bond to any other atom
- or they may be part of a molecule which may or may not be in linked to some other molecule, including one which is part of an amino acid or nucelotide chain
- or there may be several het molecules linked in a chain similar to chains of amino acids or nucleotides
This leads to the problems of connections between these atoms.
CONECT records
These records are used mainly to generate connections (bonds) between het atoms in a molecule. They use the serial number of the atoms involved, so for each bond there is a pair of atoms each of which has a unique serial number. This makes it easy to generate the bonds - just find the atoms making up each pair in the atoms array and generate a spline between the positions of those two atoms. As you might expect by now, it isn't quite that straightforward. Each CONECT record specifies what we might call a 'primary' atom and then up to four 'secondary' atoms to which the primary is connected. This is actually quite efficient but then the PDB file complicates the simplicity by specifying each bond twice, with each atom as a primary atom or as one of the secondary atoms. Parsing that to exclude duplicate bonds is difficult and slow, so I decided to accept the duplicates because it doesn't interfere with display.
Except...disulphide bonds. As we know (see part 8 of this series) these are specified by SSBOND records. That works fine. But such bonds are also given in CONECT records, so now, we have some bonds being duplicated by separate parts of the PDB file. Which wouldn't matter if it wasn't for the fact that bonds from SSBOND records are coloured yellow (by default) to highlight them, and now we run the risk of that being negated by a bond generated from a CONECT record. So in this case I decided to exclude disulphide bonds from the CONECT records, which isn't difficult but again, it means more checking through potentially long lists of atoms to avoid the duplication.
LINK records
What about bonds between different molecules? These are specified by LINK records. However, for the purpose of this plugin, we can ignore these. The reason is that CONECT records are generated from LINK records, so if we parsed LINK records and generated bonds from them it would result in duplicates of the splines generated by CONECT records. Therefore we can ignore these records because while CONECT records are needed for protein visualisation, LINK records are not.
The user interface
There are several things we need to consider from the user's point of view. The user might want to:
- not show any het atoms and/or bonds
- only show the het atoms and/or bonds
- not show specific het atoms (e.g water, which in many cases are rendered as isolated oxygen atoms since the hydrogen atoms are rarely present in the PDB file anyway)
This means that the display of het atoms must be controlled independently of atoms in amino acids or nucleotides. Since the het atoms may or may not be part of molecules which themselves are part of a chain - and there is no way to determine if that is the case - and because the het molecules can be anything, it is not possible to colour het atoms/molecules by chain or by residue, only atoms and/or bonds. Nor is is possible to represent het molecules in the ribbon format.
Results
Here are some proteins with het atoms showing what sort of display can result. I've chosen to show amino acid or nucleotide chains in ribbon format, because then it's much easier to get an idea of what the het atoms are doing.
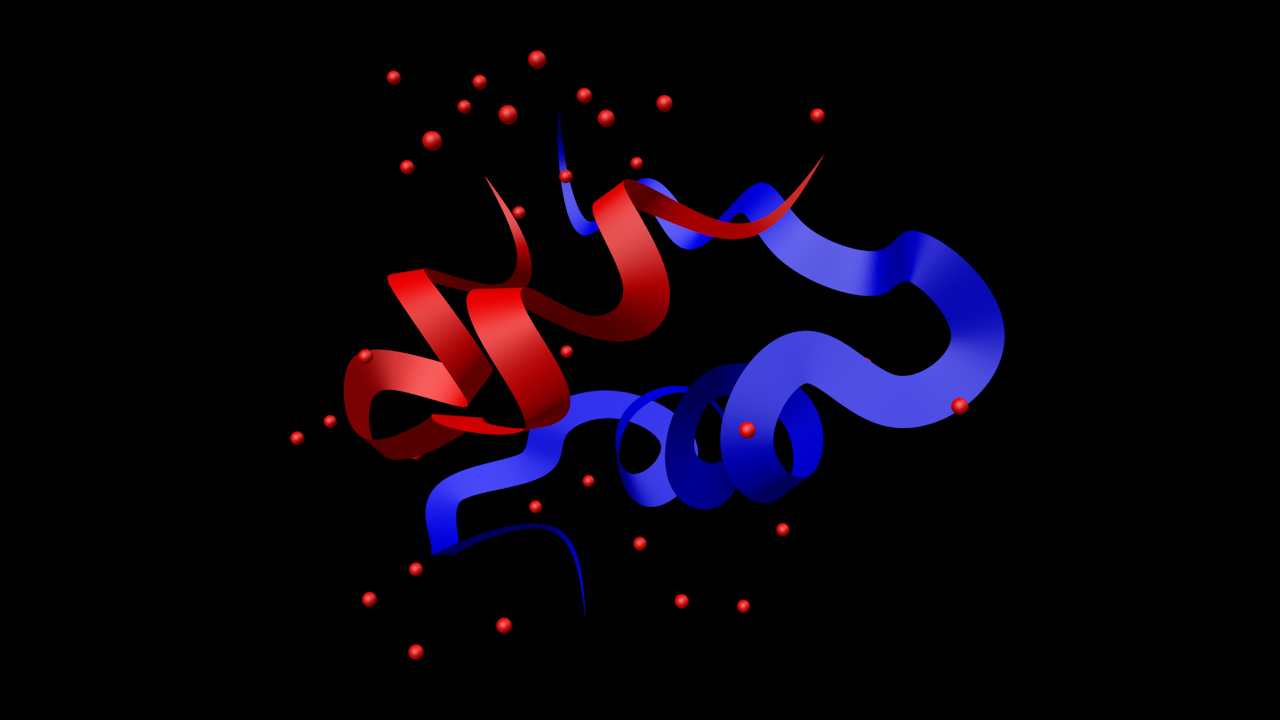
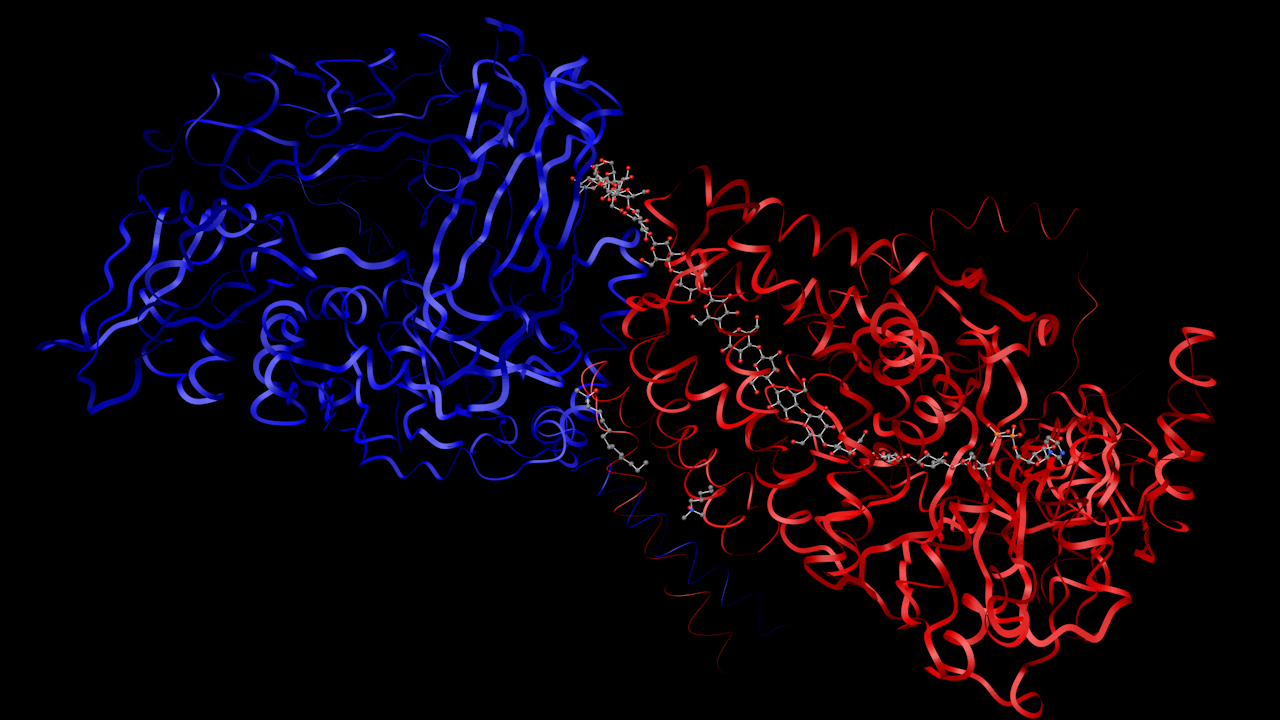
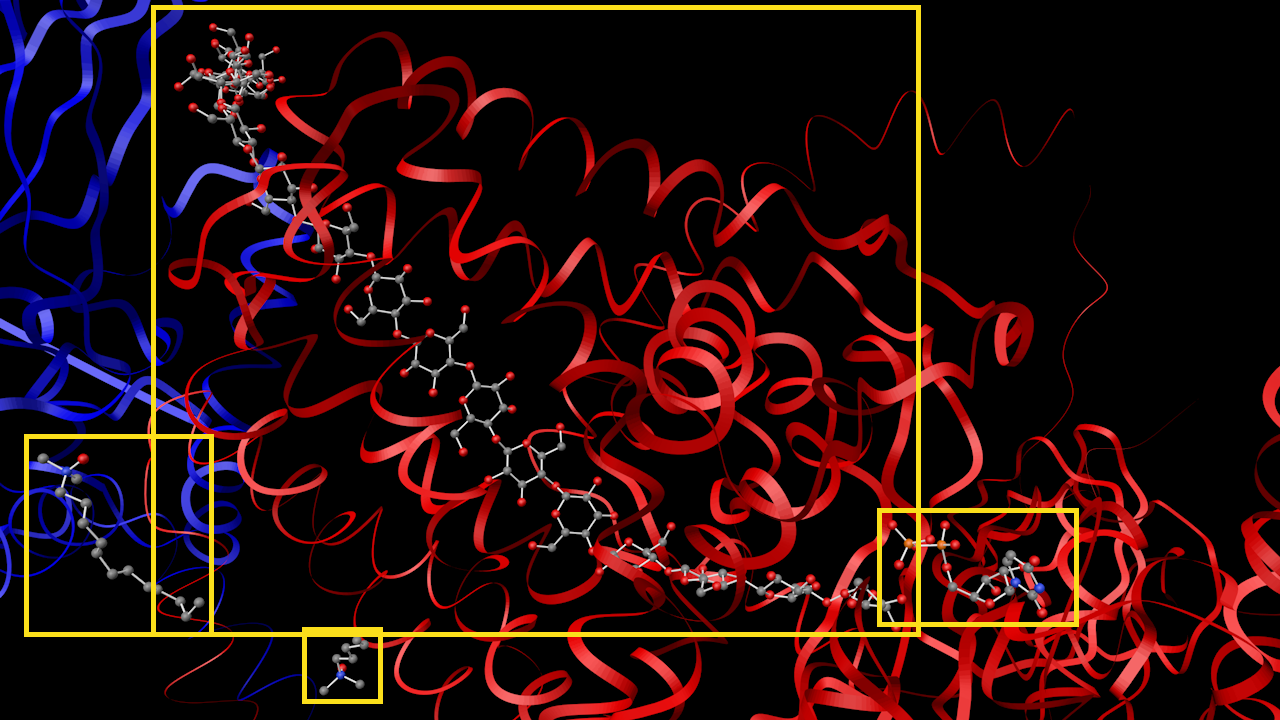
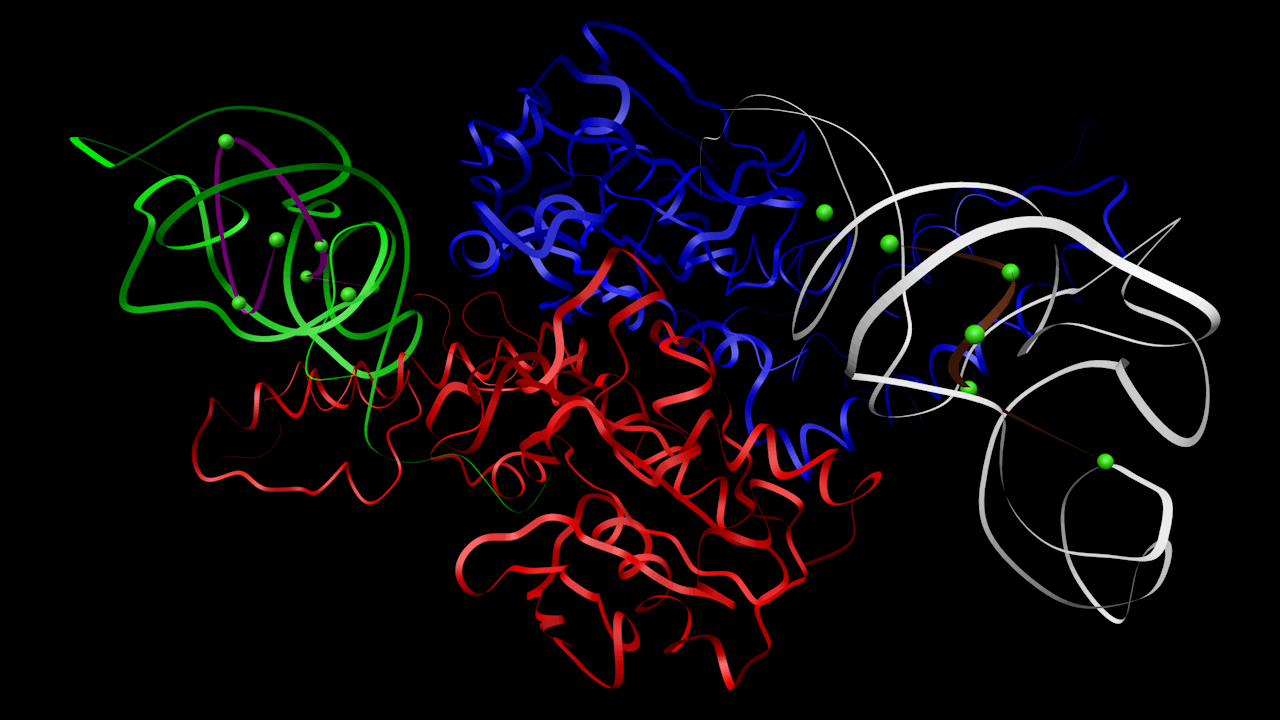
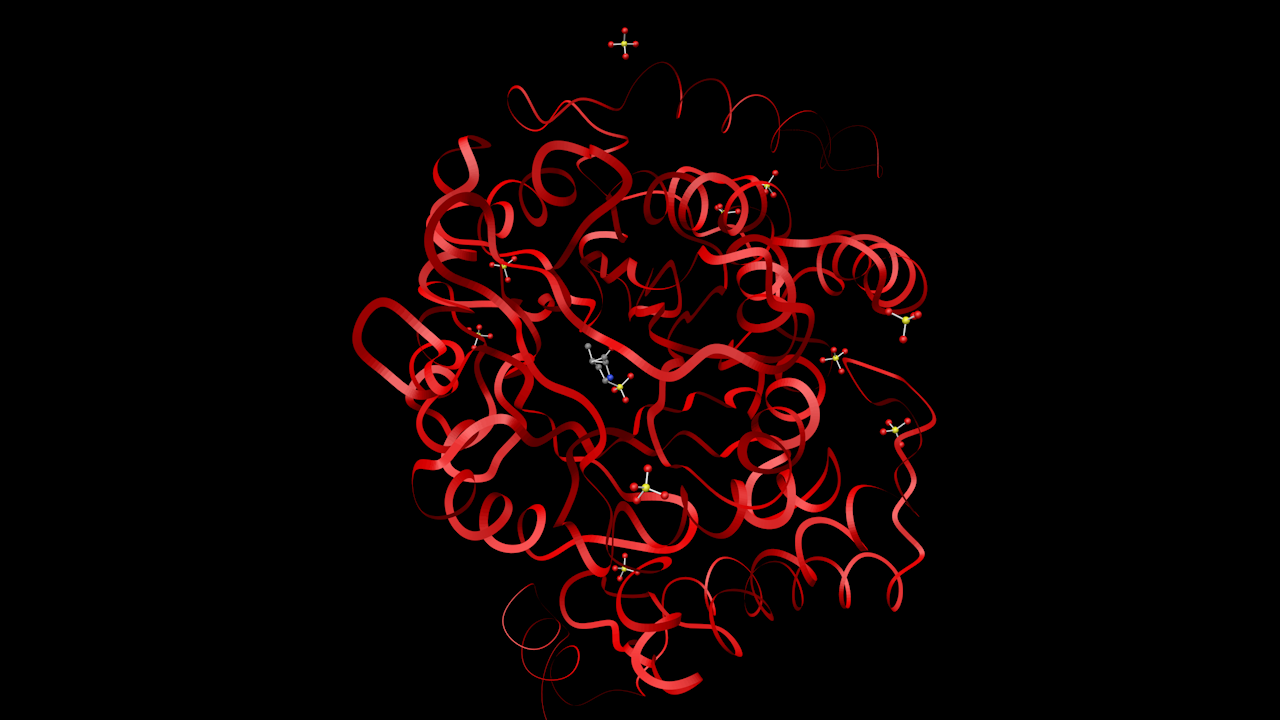
- Insulin
The red atoms are water molecules, not bonded to the main structure.
- Transferase
Several heterogen molecules can be seen, including a long chain.
- Transferase
Close up of the previous image with the heterogen molecules highlighted with yellow boxes.
- Ligase
The heterogen molecules are the green atoms, which are single calcium atoms. The bonds are not shown; some calcium atoms have bonds to the main structure, some are free-floating.
- Methyltransferase
Several heterogen atoms are shown - most are sulphate ions (the central yellow atom is the sulphur, with four oxygen atoms attached) - and with one other molecule in the centre of the ribbons.
- Signalling protein
Several heterogen molecules of two different types are scattered around the main structure.
Nearly there...
This is getting pretty close now to implementing all the features I wanted in this plugin. There are just a couple of things to add. I want to add some metadata from the protein - its title, the number of atoms present, and so on. That should be straightforward and is coming in the next section.
Page last updated July 13th 2026